 A collaborative study with my colleagues from Turin University just appeared online at the Journal of Chemical Theory and Computation. In the article “Assessment of Different Quantum Mechanical Methods for the Prediction of Structure and Cohesive Energy of Molecular Crystals” we discuss the application of different quantum chemical methods for evaluating the structure and the cohesive energy of molecular crystals. The methods range from minimal basis Hartree-Fock to local MP2, all implemented with periodic boundaries conditions in the program packages CRYSTAL and CRYSCOR.
A collaborative study with my colleagues from Turin University just appeared online at the Journal of Chemical Theory and Computation. In the article “Assessment of Different Quantum Mechanical Methods for the Prediction of Structure and Cohesive Energy of Molecular Crystals” we discuss the application of different quantum chemical methods for evaluating the structure and the cohesive energy of molecular crystals. The methods range from minimal basis Hartree-Fock to local MP2, all implemented with periodic boundaries conditions in the program packages CRYSTAL and CRYSCOR.
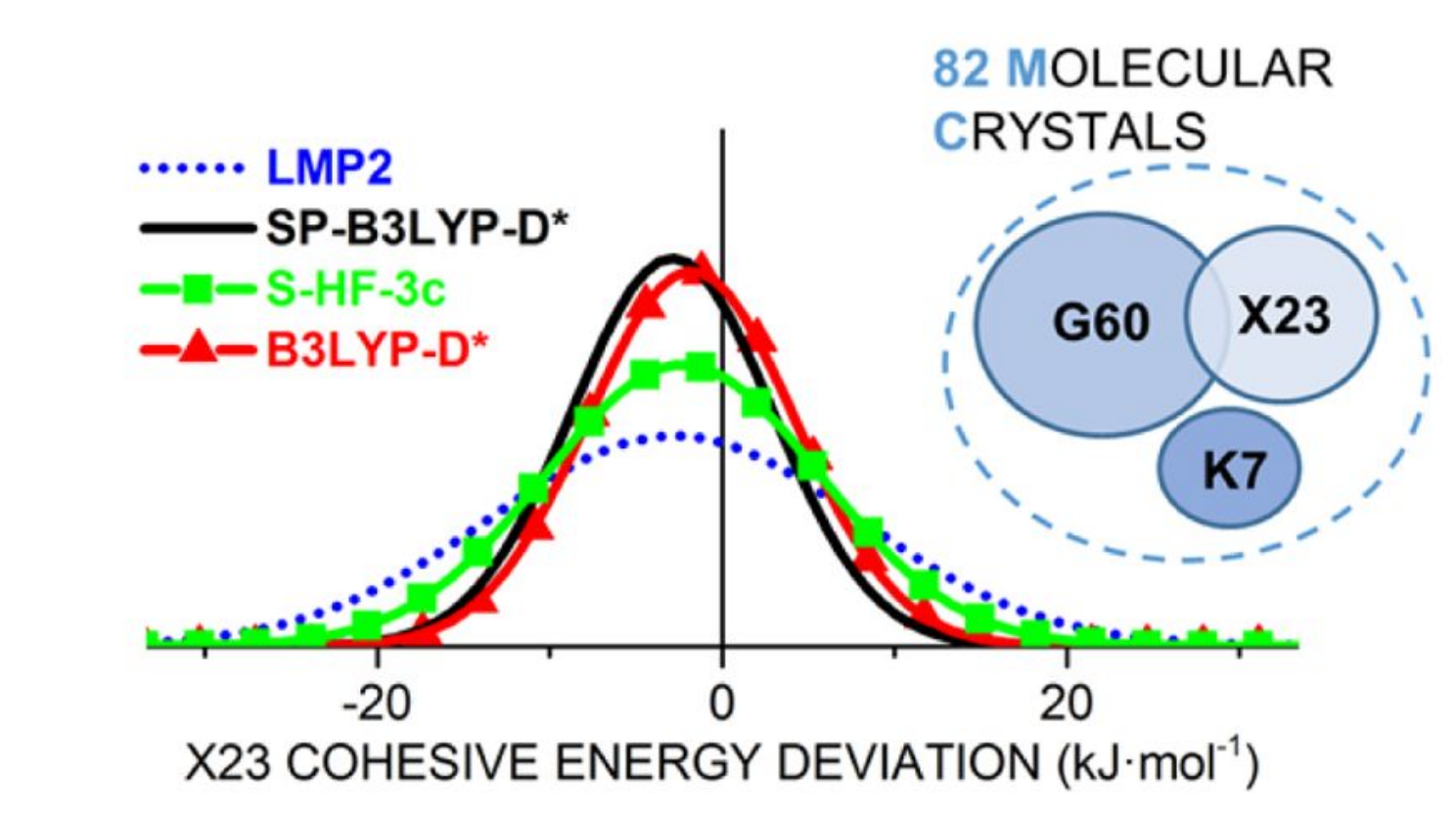
We demonstrate that the semi-empirical HF-3c method yields robust and accurate results for both energetic and geometric properties, which can be further improved by using a full hybrid functional, e.g. B3LYP-D/TZ. The LMP2 perturbation theory is applied for the first time on a diverse molecular crystal benchmark set, where basis set issues and intrinsic MP2 problems are highlighted. Find the article at JCTC.


Pingback:Psi-k Highlight of the Week - Dr. Jan Gerit Brandenburg