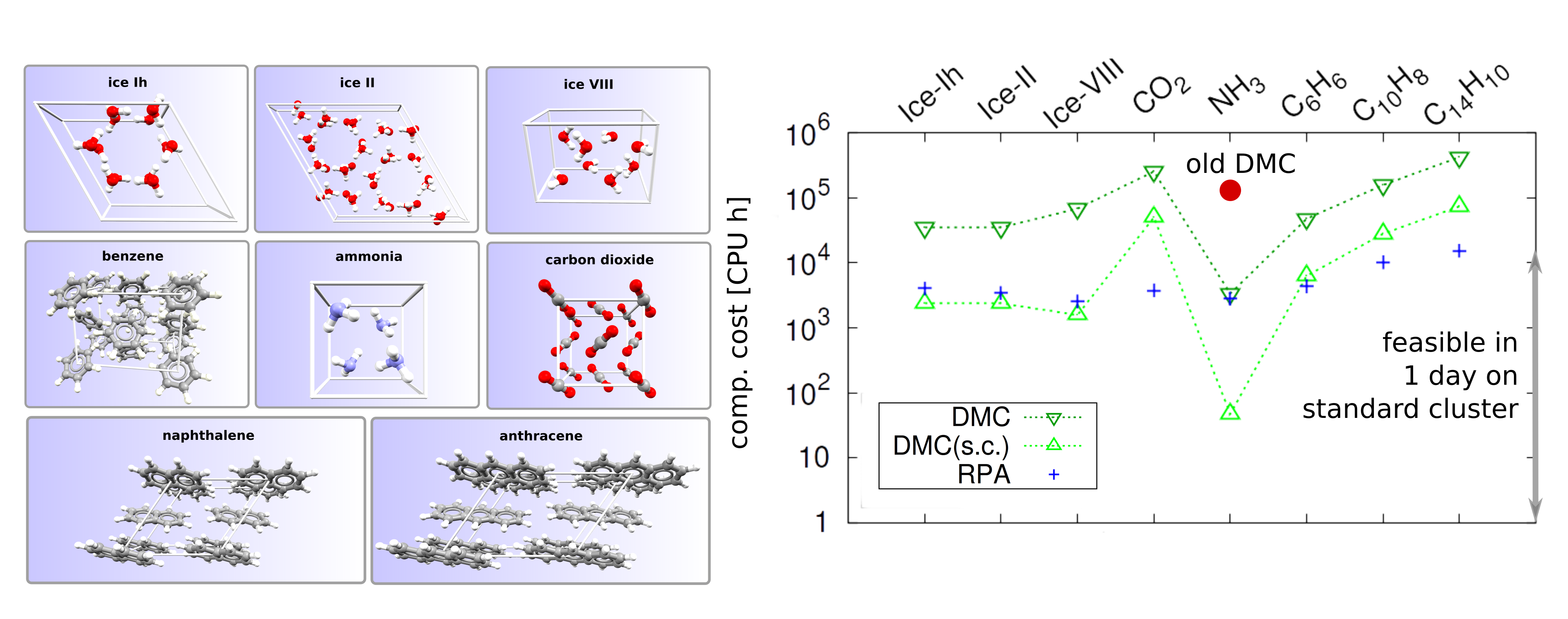
Our recent work on “surprisingly affordable” quantum Monte-Carlo for molecular crystals has been published: Proc. Natl. Acad. Sci. U.S.A, 2018, 115, 1724.
PNAS Significance statement:
“Computational approaches based on the fundamental laws of quantum mechanics are now integral to almost all materials design initiatives in academia and industry. If computational materials science is genuinely going to deliver on its promises, then an electronic structure method with consistently high accuracy is urgently needed. We show that, thanks to recent algorithmic advances and the strategy developed in our manuscript, quantum Monte Carlo yields extremely accurate predictions for the lattice energies of materials at a surprisingly modest computational cost. It is thus no longer a technique that requires a world-leading computational facility to obtain meaningful results. While we focus on molecular crystals, the significance of our findings extends to all classes of materials.”


Pingback:water at molecular scale - Dr. Jan Gerit Brandenburg