 Our review on Simplified DFT methods for consistent structures and energies of large systems has just been published:
Our review on Simplified DFT methods for consistent structures and energies of large systems has just been published:
J. Phys.: Condens. Matter 2018, 30, 213001.
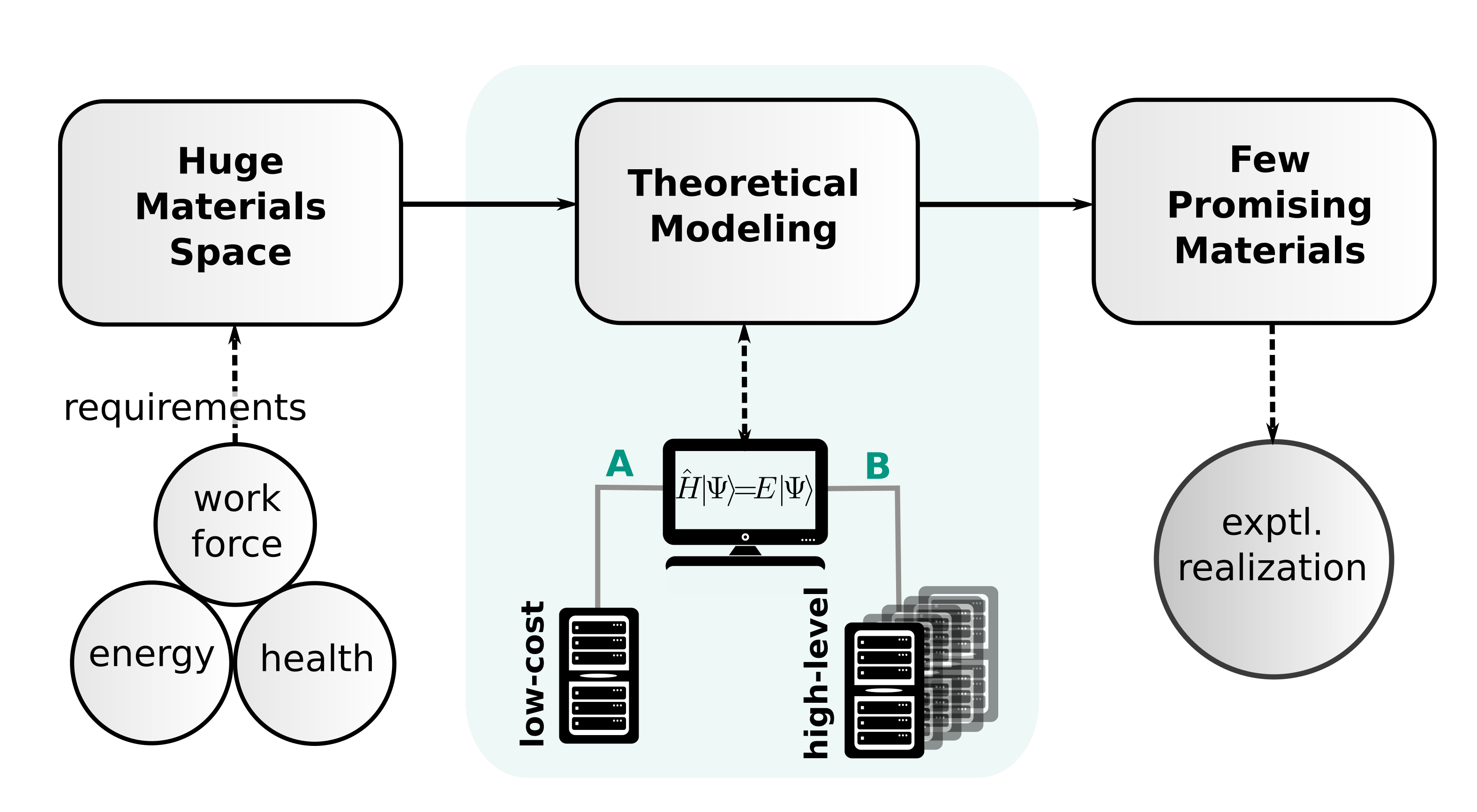
Even though affordable many-body electronic structure methods emerge, we still see DFT as an irreplaceable tool for (a) the routine calculation of structures and properties of systems with medium size of about 100 atoms and (b) the electronic structure description of increasingly large systems with well above 1000 atoms. In this topical review, we presented a set of low-cost methods and mainly focused on the electronic structure part by combining compact orbital basis sets with semi-classical correction potentials. Substantial speed-ups of one to three orders of magnitude can be achieved while keeping the good DFT-D accuracy.
This hierarchy of methods is well suited for the every-day calculation on systems of modest to large size and expect a significant impact on the crystal structure prediction algorithms and on large scale material screenings in general.
Simplified Density Functional Approximations